EMIL
CAT Endstation
The CAT laboratory operated by the Fritz-Haber-Institut (FHI) and the Max Planck Institute for Chemical Energy Conversion (MPI-CEC) is dedicated to in-situ and operando spectroscopy on functional materials, focusing on catalysis. The aim of the instrumentation is to provide the users a state of the art, quantitative molecular-level description of chemical interfaces that are of high relevance for a detailed understanding of the working principle of catalysts. The installed Ambient Pressure X-ray photoelectron spectroscopy (AP-XPS) spectrometer is used for in-situ/operando interrogation of electronic structure and chemical composition of catalytically active gas/solid and liquid/solid interfaces. Buried interfaces become accessible by the high kinetic energy capability of the electron spectrometer and the very broad photon energy range provided by the EMIL beamlines.
The CAT endstation is located at the 2nd combined focus of the EMIL beamline cluster served both by UE48 and U17 sources. The general layout is presented as Figure 1. A continuous photon energy range from 80 eV – 10000 eV is available at CAT

Figure 1: Drawing of the refocusing mirror vacuum chambers “M4-soft” and “M4-tender”, the differentially pumped interface section labelled “Diff 1”, “Diff 2”, “Diff 3” and the Ambient-Pressure XPS (AP-XPS) spectrometer with an experiment module attached in the combined focus of the soft X-ray beam delivered by the UE48 undulator and the hard X-ray beam delivered by the U17 undulator. The vacuum tubes for the soft and hard beam are indicated in lilac and red color, respectively
The core of the CAT endstation consists of a commercial AP-XPS spectrometer (SPECS PHOIBOS 150 NAP-XPS) and a homemade XPS chamber equipped with a versatile multi-purpose manipulator providing several electric contacts, fluid dosing lines and the option for Near Infrared (NIR) laser heating. The electron spectrometer covers the kinetic energy range up to 7 keV. To cope with the broad range of samples and reaction conditions, a highly modular instrumentation approach with standardized interfaces on different levels is realized at the CAT endstation. Details of the spectrometer, the modularity approach including various suitable reaction cell concepts can be found in the literature and references therein(Bluhm, Hävecker et al. 2004). This approach facilitates an easy adaptation of the station to the demands of the users while keeping the spectrometer aligned and optimized with the beamline. It is noteworthy to mention that the equipment used at CAT is compatible and retrofits to the MPG stations at ISISS and BElChem beamlines. The differentially pumped interface to the beamline allows a time-efficient operation of the CAT endstation as it avoids downtimes to establish high vacuum conditions before the synchrotron measurements can continue after an adaptation to user needs of the set-up. This interface section holds units for beam diagnostics and provides space for beam filter and attenuators, photon energy calibration tools, reference foils etc. The differentially pumped section could easily be upgraded to facilitated windowless operation of the AP-XPS instrument without a beam entrance membrane (SiNx, 75nm thickness) that is currently installed to separate the vacuum conditions of the beamline from the gas in the XPS chamber. The unique laboratory infrastructure implemented at CAT complements the endstation. It allows handling of ultra-clean gases to flammable, toxic and corrosive fluids. Additionally, CAT strongly benefits from being embedded in the extensive EMIL infrastructure like on-site access to the EMIL chemistry laboratory and sample characterization methodologies installed at SCALA. Table 1 points out some of the versatile key features of the CAT endstation at EMIL including the essential combination of spectroscopy with high performance product analysis as a prerequisite for meaningful in-situ/operando spectroscopy.
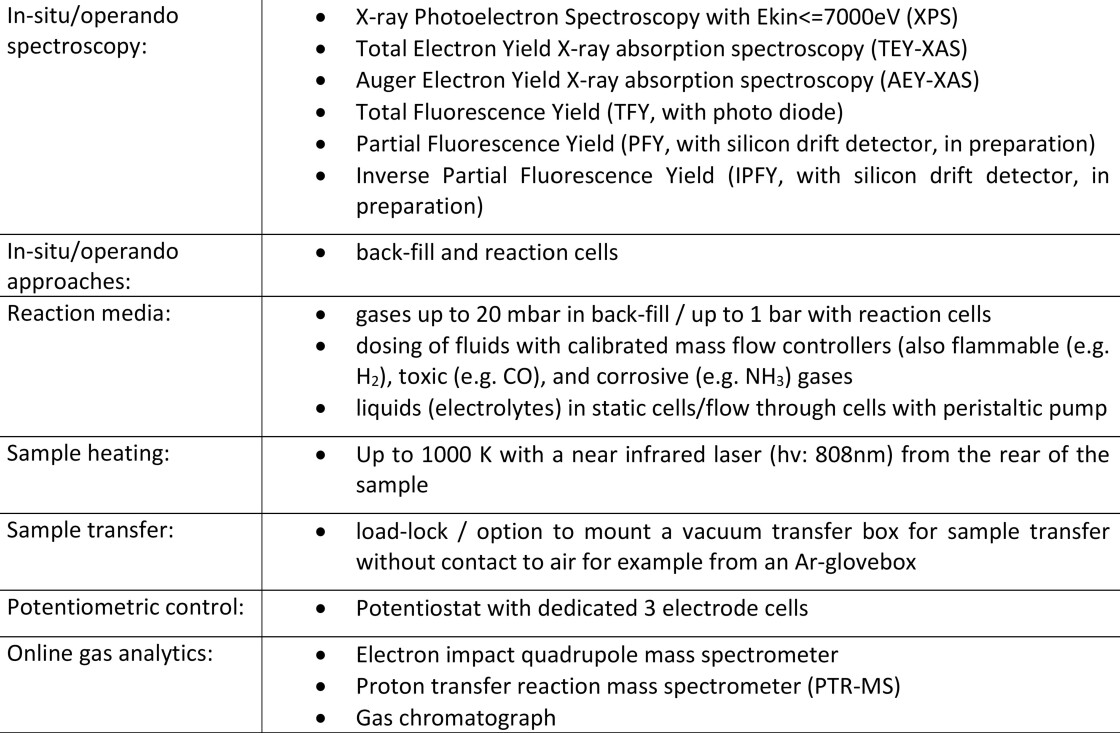
Table 1 Key methodologies and experiment condition that are realized at CAT.
The following example study of the selective hydrogenation reaction of acetylene on a Pd catalyst exemplifies some of the capabilities at CAT. The operando investigation of this catalytic reaction over a large range of pressures, bridging the pressure gap between NAP conditions in the low mbar range up to higher (atmospheric) pressures is described.
Palladium is an important catalyst for hydrogenation reactions due to its high conversion and selectivity. One important example is the hydrogenation of alkynes to alkenes.(Borodziński and Bond 2006). There are two important phases involved in this reaction: the bulk β-palladium hydride is considered as the unselective phase while carbon located at interstitial subsurface sites causes the high catalytic selectivity.(Teschner, Borsodi et al. 2008). The modular approach at CAT offers the capabilities to investigate the Pd catalysis over a broad range of experimental conditions. The investigation of the same sample by AP-XPS and AP-XAS offers new insides in the interplay of the involved phases as well as in the electronic structure of occupied (XPS) and unoccupied (XAS) states.
Operando AP-XPS investigations have been performed in a module for gas phase heterogeneous catalysis in the back-fill mode. To achieve high surface sensitivity soft X-rays with 830 and 490 eV photon energy from the UE48-PGM branch were used. The sample was mounted on a sapphire support and placed in front of the spectrometer nozzle, at the intersection of spectrometer and (both soft and tender) X-ray beam focal point. The chamber was backfilled with the reaction gas mixture and the product analysis was done with a Thermo Scientific TRACE 1300 gas chromatograph using a FID (flame ionization detector) detector. The sample was annealed by illumination with an IR laser (λ = 808 nm) from the rear side. The Pd catalyst was prepared by electrodeposition on a carbon coated quartz substrate (0.0 V vs Ag/AgCl, 1 mM PdCl2 in 0.1 M HCl, 5 mC, about 6 nm nominal thickness).

Figure 2: a) Pd 3d5/2 AP-XPS spectra of electrodeposited Pd during cleaning and selective hydrogenation reaction (black). The components for the deconvolution are metallic Pd (dark blue), Pd-C (light blue), surface oxide (orange), PdO (red) and the envelope (green). b) Pd L3 XANES spectra. Top: from the same sample as in a). Bottom: electrodeposited Pd in a high pressure cell. c) Ethylene selectivity (red) of the operando measurements shown in a) and b), and normalized intensity of the hydride peak obtained from the NEXAFS spectra shown in b).
Fig. 2 a) shows the corresponding Pd 3d5/2 spectra. The spectrum during an oxygen cleaning (200°C, 0.2 mbar O2) shows mainly metallic Pd (dark blue, 334.9 eV BE) together with the characteristic doublet(Lundgren, Kresse et al. 2002, Zemlyanov, Aszalos-Kiss et al. 2006) at 335.4 and 336.2 eV BE (orange) for a surface oxide layer. No significant bulk oxidation occurs at these conditions as the corresponding signal for PdO(Gabasch, Unterberger et al. 2006) at 336.6 eV (red) has only a very minor contribution. When applying reaction conditions (125°C, 1 mbar H2:C2H2=5:1) the peak shape changes strongly. It is now dominated by a peak at 335.6 eV (light blue) which can be attributed to Pd in contact with carbon and thus the interstitial carbon phase(Teschner, Borsodi et al. 2008). In a depth profiling the relative intensity of the carbon related species increases from 72.4% for hv=830 eV to 81.9% for the more surface sensitive hv=490 eV. In addition, the quantification (for hv=830 eV) gives a thickness of this phase of about 5 ML. The combination of an enrichment at the surface and a thickness of several monolayers proves that the majority of its contribution comes from the subsurface region of the catalyst. The catalytic performance during the XPS measurement showed a high ethylene selectivity of 97.1%.
The corresponding operando XAS analysis was done using the U17-DCM branch and the Pd L3 spectra are shown in Figure 2 b). The spectra in the upper part of Fig. 2 b) were taken at the same sample as for the AP-XPS analysis. The absorption signal was collected by biasing the spectrometer nozzle to 90V to attract the (secondary) electrons and measuring their current (total electron yield, TEY). The peak shape obtained during the mild oxidation step (red) is the characteristic one for metallic palladium, with a white line position at 3173.5 eV (Tew, Janousch et al. 2011). The surface oxide is not visible here due to the larger information depth of TEY-XAS compared to low kinetic energy XPS. The spectrum after the selective hydrogenation reaction measured in high vacuum and at room temperature is shown in black. The broadening of the white line towards higher energies indicates the formation an interstitial carbon phase (Tew, Janousch et al. 2011) which is in line with the XPS results.
With the back filling mode, the necessary H2 partial pressure to trigger the formation of the β-hydride phase could not be reached. To overcome this limitation, measurements were performed in a high pressure XAS cell, which can be easily mounted to the setup due to the modular approach. The description of the cell is given in Refs. (Velasco-Vélez, Pfeifer et al. 2016, Velasco-Vélez, Chuang et al. 2020). In short, the cell is closed by a 100 nm SiNx window, coated with a 3 nm Cr adhesion layer followed by 20 nm PVD Au. The Pd sample was prepared on the inner side by electrodeposition (-0.5 V vs. Ag/AgCl, 1mM PdCl2 in 0.1 M HCl for 20 min, about 9 nm nominal thickness). As the SiNx window is transparent for X-rays, but not for electrons, XAS was measured via total fluorescence yield (TFY) by using a photodiode. The reaction mixture (H2:C2H2=5:1, balanced with He) was dosed at room temperature while the product analysis was achieved with GC. The spectrum taken at 18 mbar shows a pure metallic Pd shape. The absence of the carbon related phase could be explained either by an instability of this phase at this pressure or by the higher bulk sensitivity of TFY compared to TEY. With increasing pressure there is a change in peak shape between 18 and 36 mbar. The new peak shape has a shifted white line to 3174.4 eV and a new peak around 3180 eV, which is the fingerprint of the β-hydride formation (Davoli, Marcelli et al. 1989). The observed pressure range for the transition fits well to the reported thermodynamic stability of the β-hydride phase at room temperature (Frieske and Wicke 1973) as well as to XRD (Velasco-Vélez, Teschner et al. 2018) and EXAFS (Bugaev, Guda et al. 2017) reports.
The catalytic performance during these measurements is summarized in Fig. 2 c) by showing the selectivity towards ethylene (red dots) and the normalized height of the hydride related peak at 3180 eV (black dots). The selectivity drops at higher pressures than 1 mbar might be related to the missing interstitial carbon phases, although effects of different temperature and sample cannot be excluded. With further increasing pressure the selectivity drops further which could be related to the presence of the β-hydride phase that had been reported to be unselective (Velasco-Vélez, Teschner et al. 2018). On the other hand, just after the formation of the hydride phase the selectively remains quite constant, and mainly drops at higher pressures, indication a more complex behavior of the sample, indicated by a slight increase of the hydride peak intensity between 36 mbar and 120 mbar until it reaches saturation.