Protein crystallography at BESSY II: a tool for decoding the building blocks of life

Over 4.000 structures have been decoded at BESSY II (as of Dezember 2022), here you can get a picture of them
BESSY II is a brilliant light source for research, with soft and tender X-rays. With its special synchrotron light, we study proteins that play a crucial role in the functioning of viruses, bacteria, and cells, for example.
Protein molecules are involved in nearly all biochemical processes of living organisms and viruses. A single macromolecule can be made up of hundreds of amino acids, forming a huge and highly complex three-dimensional architecture. Parts of these macromolecules are folded, other parts look like loops or spirals, and there can be branches and symmetries. This 3D architecture determines how a protein functions.
How does protein crystallography work?

The picture shows so-called BphA4 protein crystals. BphA4 is a subunit of a bacterial protein.
© Tajana Barthel (HZB MX)
At BESSY II, the 3D structures of proteins are decoded at three beamlines using X-ray diffraction. These beamlines are also known as MX beamlines. The abbreviation MX comes from the English, where M stands for "Macromolecular" and X for "X-ray structure analysis".
First, the proteins crystallise out of the solution - a challenging process in which a special sample holder from the HZB MX group facilitates the handling of the tiny and sensitive crystals. The samples are then cooled in liquid nitrogen and automatically placed in the X-ray beam to create a diffraction pattern. Sophisticated algorithms are used to analyse this pattern in order to reconstruct the proteins in three dimensions together with their electron density, which provides important information on binding and chemical properties.
The MX team at BESSY II

HZB MX-Team (May 2024)
The Joint Research Group Macromolecular Crystallography, or MX team for short, has grown a lot over the years. It now numbers 14 people..
Two MX beamlines runs completely via remote access. This means the users send in their samples to us, the team prepares them for measurement, and the rest is done by the users in front of their computers. They can control (almost) everything from there.
Portrait Manfred Weiss (Head MX-Team)
Protein crystallography and the coronavirus
Which protein of the SARS-CoV-2 virus was investigated at BESSY II?
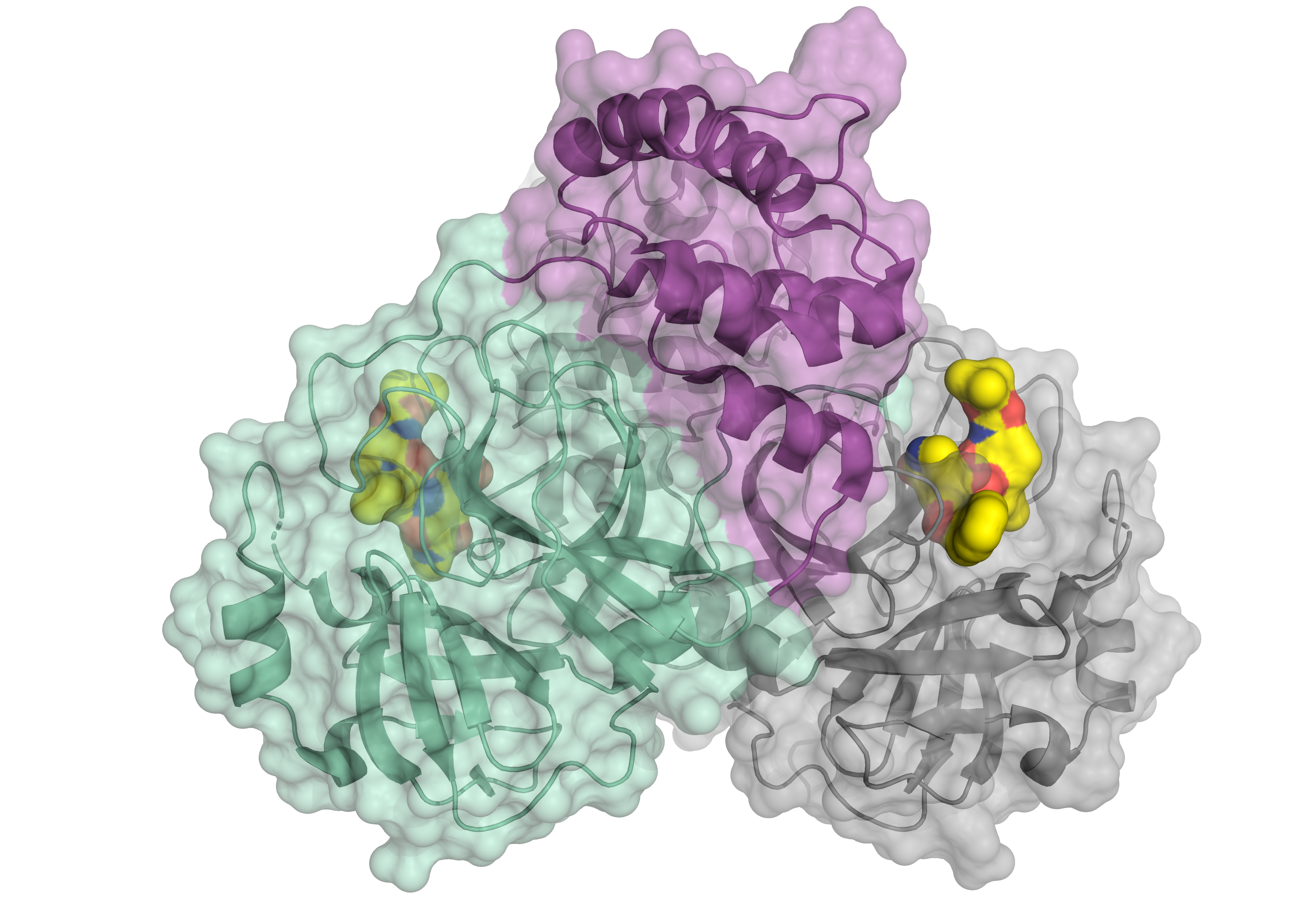
Schematic representation of the corona virus protease. The enzyme occurs as a dimer consisting of two identical molecules. One part of the dimer is shown in green and violet, the other in grey. The small molecule in yellow binds to the active centre of the protease and might serve as a blueprint for an inhibitor. © HZB
The main protease of the SARS-CoV-2 virus was studied at BESSY II. This protein is essential for the reproduction of the virus. The chemist Prof. Dr. Rolf Hilgenfeld carried out these investigations on BESSY II in order to identify suitable docking targets for an active agent, such as an inhibitor.
Why is protein crystallography so important for drug development?
An active substance must “dock” (chemically bind) to a location on the protein. Like a key in a lock, it must fit in order to inhibit the function of the protein. Many proteins are being studied at BESSY II with the aim of developing active agents.
A high-throughput method is available at the MX beamlines for this purpose, allowing hundreds of drug fragments to be systematically tested in protein crystals. Effective therapeutic drugs can be developed much more quickly using these fragments as building blocks for active substances.
Film: A team from the University zu Lübeck in Germany has decoded the main protease of the coronavirus using X-ray light from BESSY II.
01:11